- El equilibrio de desnaturalización
- Medida de la estabilidad conformacional
- Patrón termodinámico de la estabilidad proteica
- Interacciones que contribuyen a la estabilidad e importancia relativa
- Experimento de Anfinsen y paradoja de Levinthal
- Medida de la velocidad de plegamiento
- Modelos clásicos del plegamiento y especies observadas
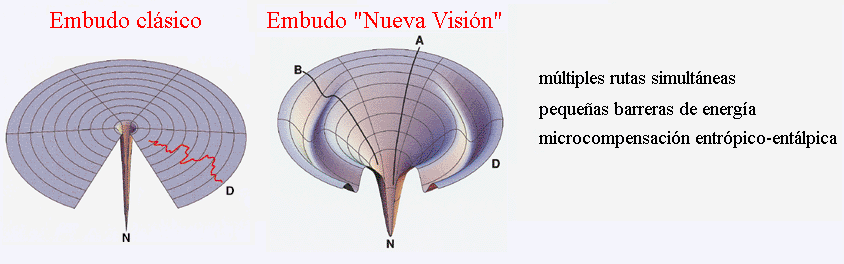
- La ´nueva visión´ del plegamiento
- Plegamiento in vivo y chaperonas

Nobel Prize in Chemistry 1972 to Christian B. Anfinsen
"For his work on ribonuclease, especially concerning the image002connection between the amino acid sequence and the biologically active conformation"
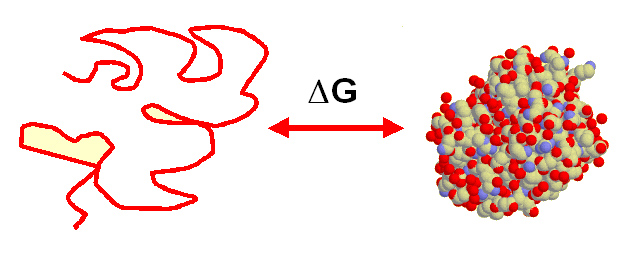
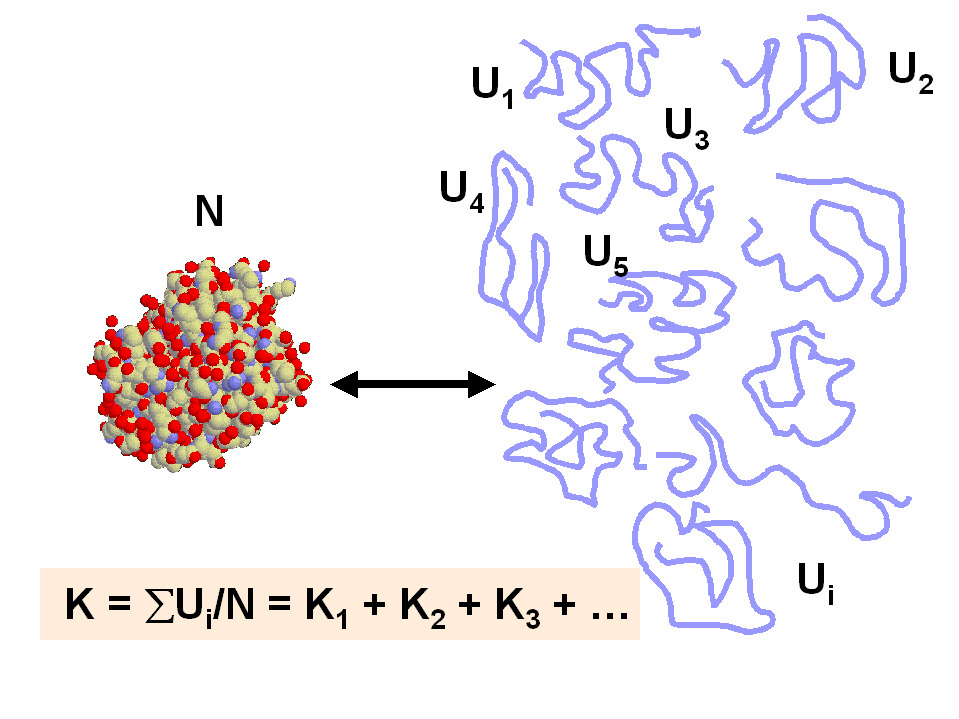
- El equilibrio de desnaturalización. La conformación nativa (plegada) de las proteínas se encuentra en equilibrio con el estado desnaturalizado (desplegado). La estabilidad conformacional de una proteína es la diferencia de energía libre estándar del equilibrio. El estado desnaturalizado es, en realidad, un conjunto de numerosas conformaciones desplegadas de similar energía.
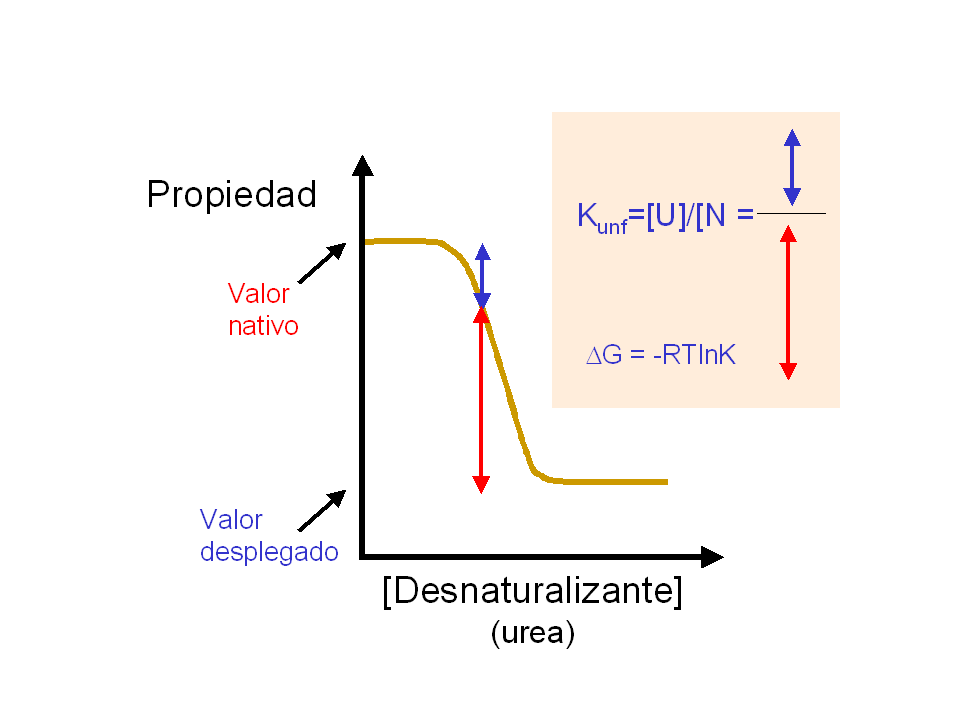
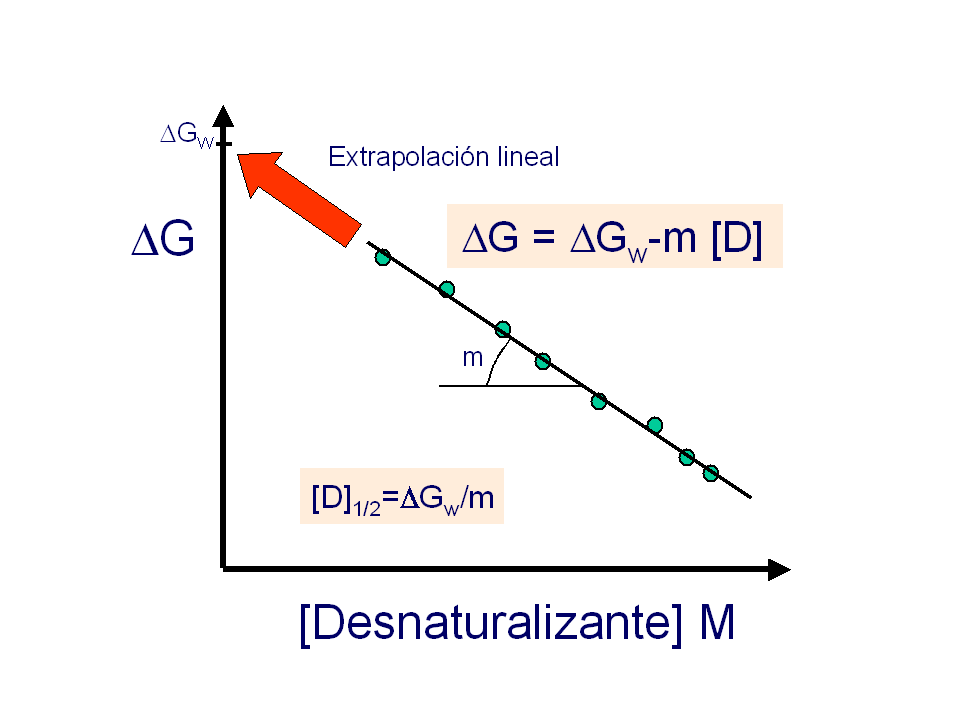
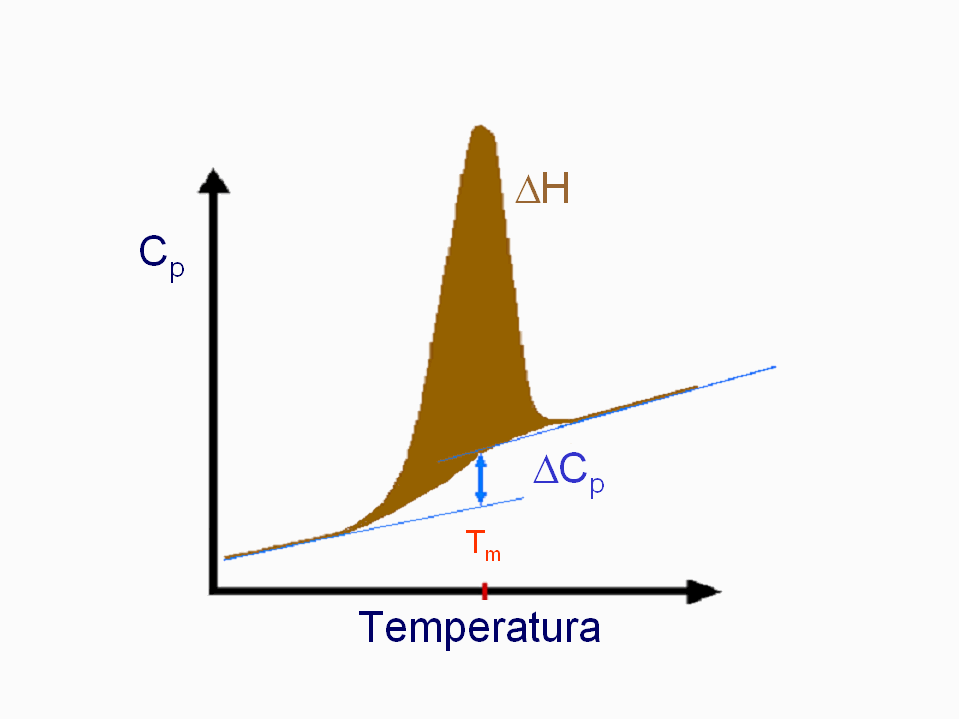
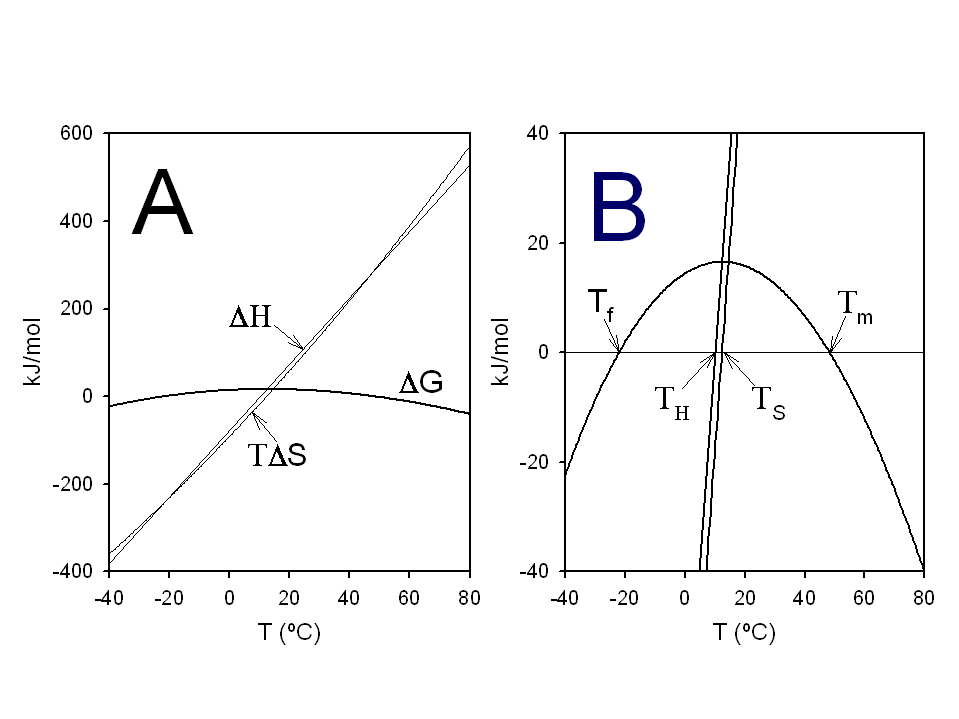
- Medida de la estabilidad conformacional. Las diferencias de energía libre se calculan siempre a partir de constantes de equilibrio. La forma más sencilla de medir la constante del equilibrio de plegamiento es el método de extrapolación lineal (LEM) que se basa en medir una propiedad espectroscópica de la proteína (fluorescencia, absorbancia, dicroísmo circular) a distintas concentraciones de una sustancia desnaturalizante (p.e.: urea) y extrapolar la estabilidad a concentración nula de desnaturalizante. Típicamente, la estabilidad de las proteínas es muy pequeña: varía entre -5 y -15 kcalmol-1. ¿Qué % de moléculas está plegado si ΔG=-5 kcalmol-1? La estabilidad se puede medir también mediante calorimetría diferencial de barrido (DSC). A partir de la diferencia de entalpía (ΔH), la diferencia de capacidad calorífica (ΔCp) y la temperatura media de desnaturalización (Tm), la estabilidad se puede calcular a cualquier temperatura. La curva de estabilidad en función de la temperatura indica que las proteínas se desnaturalizan por calor y por frío.
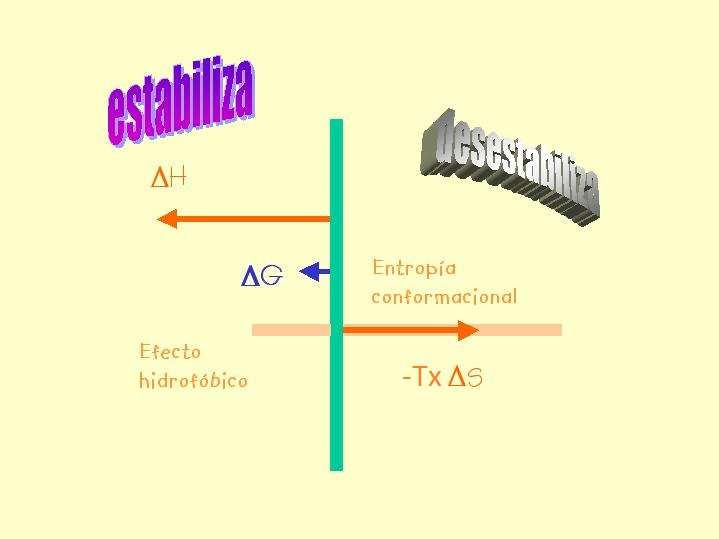
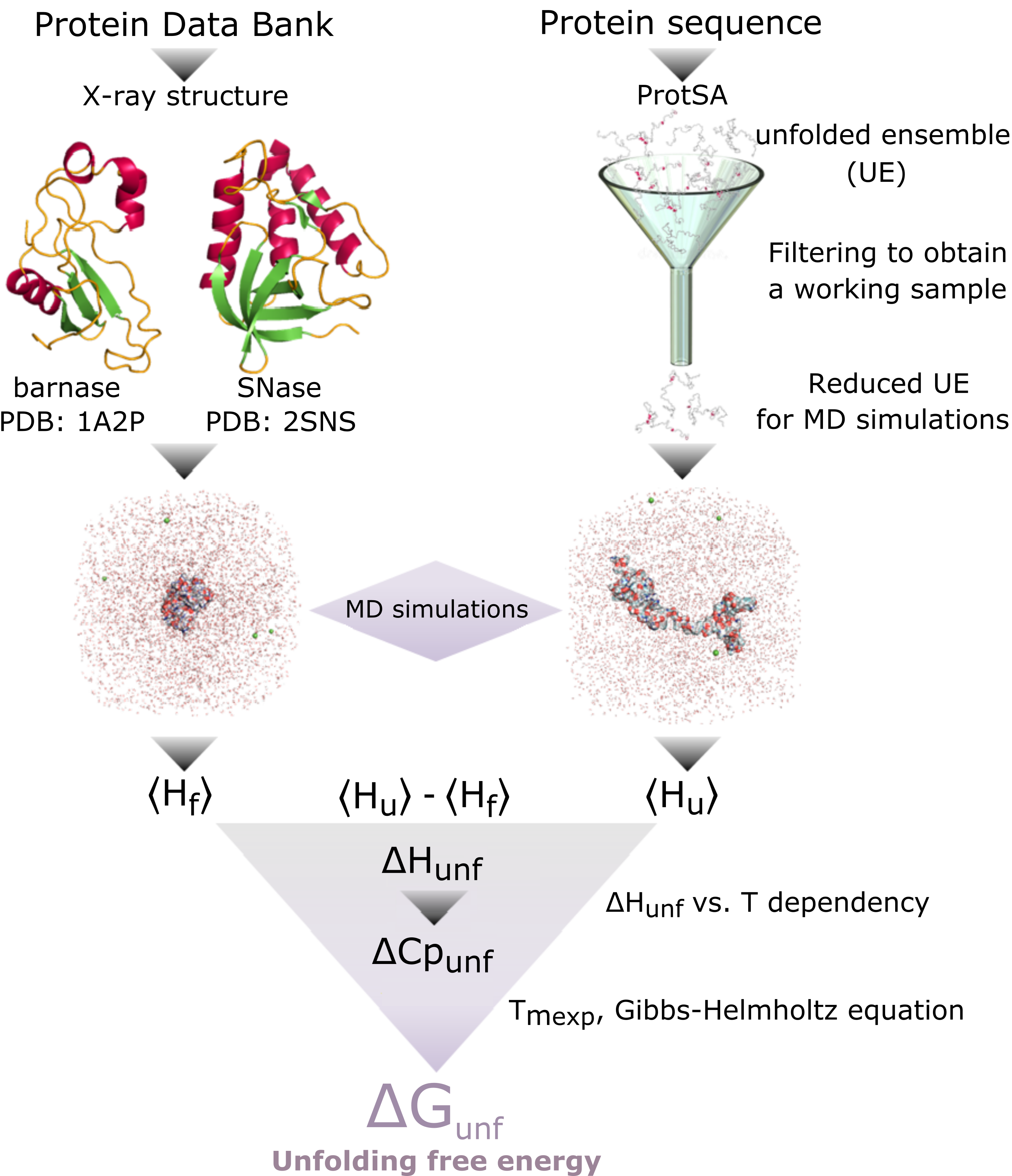
- Patrón termodinámico de la estabilidad proteica. A temperaturas próximas a la de desnaturalización, la diferencia de entalpía del plegamiento presenta valores de entre –100 y -300 kcal mol-1. De acuerdo al segundo principio de la Termodinámica esto implica que el cambio de entropía neto del equilibrio de plegamiento es negativo (-TΔS positivo, y de valor absoluto próximo al de ΔH). Al plegarse la proteína la entropía de la cadena polipeptídica disminuye mucho y la del agua aumenta (pero menos; efecto hidrofóbico). La estabilidad de las proteínas es pequeña porque la estabilización entálpica está casi compensada por la desestabilización entrópica global. En principio, la estabilidad de las proteínas podría calcularse restando las interacciones entre átomos de la proteína y del agua en el estado desnaturalizado de las interacciones en el estado nativo pero haría falta mucha precisión. Recientemente se ha conseguido calcular ΔHu, calculando previamente Hu y Hf (a partir de simulaciones de dinámica molecular) y restando. También se ha conseguido calcular ΔCp como la pendiente de una representación de ΔH calculada a tres temperaturas frente a dichas temperaturas.
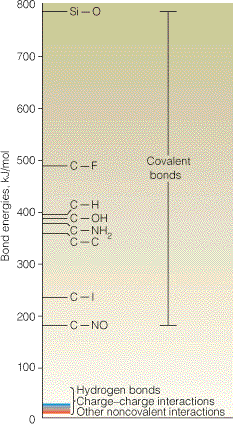
- Interacciones que contribuyen a la estabilidad. importancia relativa. Las interacciones que se observan en el estado nativo son de los siguientes tipos:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
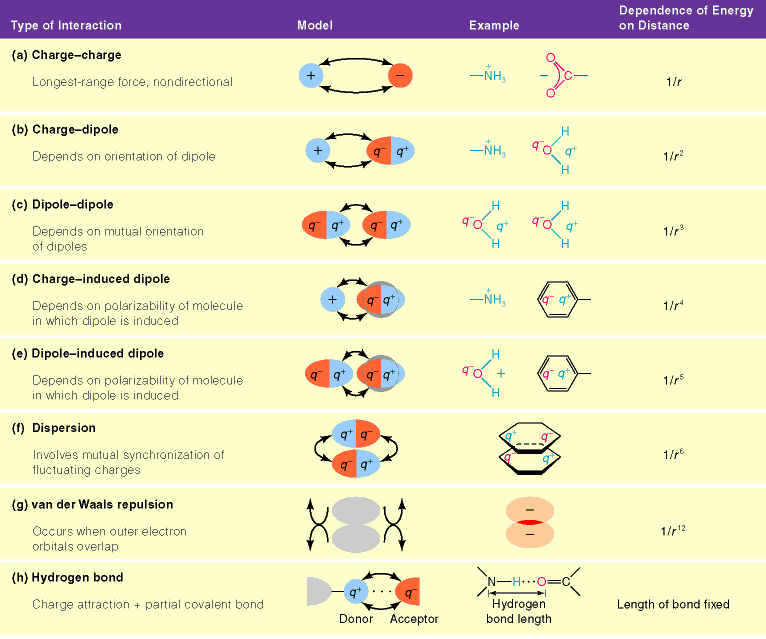
- Carga/carga: (residuos ionizables). Obedecen la Ley de Coulomb. Según se distribuyan las cargas en la proteína, su efecto conjunto puede ser estabilizante o desestabilizante.
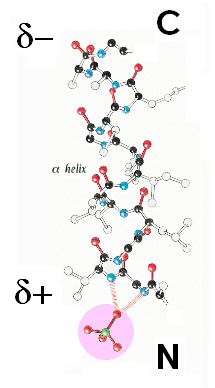
- Carga/dipolo: Un ejemplo es el de residuos cargados positivamente con el dipolo de una hélice alfa en el extremo C y residuos negativos con el dipolo del extremo N. Muchos metabolitos fosforilados se unen a extremos N de hélices alfa.
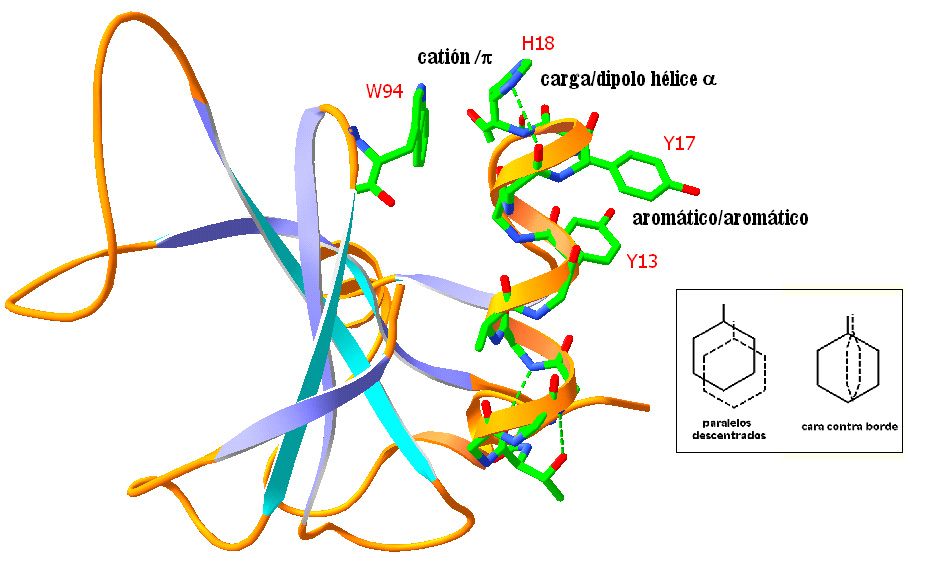
- Carga/cuadrupolo: Interacciones catión/pi entre residuos cargados positivamente y nubes π de anillos aromáticos. También se dan en sitios de unión de cationes (p.e.: en canales iónicos).
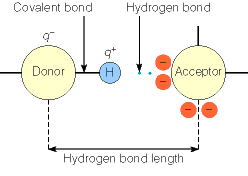
- Dipolo/dipolo. Un ejemplo notable son los puentes de hidrógeno (aunque además poseen un cierto carácter de enlace covalente). La orientación más estable es con los tres átomos en línea recta.
- Cuadrupolo/cuadrupolo: Interacciones entre anillos aromáticos.
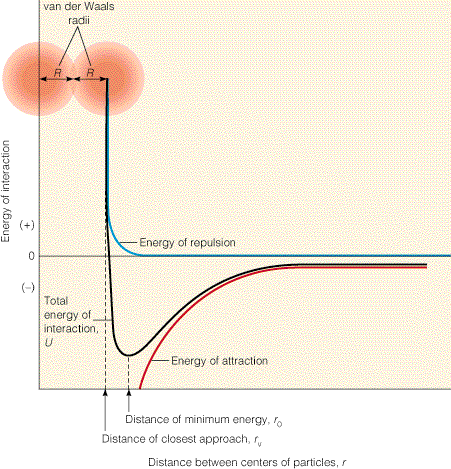
- Dipolo inducido/dipolo inducido (fuerzas de dispersión). Atraen a dos átomos cualesquiera hasta que se acercan tanto que comienzan las repulsiones electrónicas. Las fuerzas atractivas de dispersión junto con las repulsivas electrónicas se conocen como fuerzas de Van der Waals y tienen una dependencia de la distancia característica.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
El signo negativo de ΔH de plegamiento indica que algunas de estas interacciones son más fuertes en el estado nativo que en el desplegado pero no es sencillo determinar experimentalmente cuales. Hay que tener presente que la formación de interacciones en el estado nativo es siempre a expensas de la rotura de interacciones previas (intraproteicas o con el disolvente) en el estado desplegado. Hoy se piensa que las fuerzas de Van der Waals son estabilizantes porque el estado nativo está muy bien empaquetado. Respecto a los puentes de hidrógeno no hay opinión unánime (aunque es seguro que determinan la estructura proteica: estructura secundaria). Las otras interacciones son menos numerosas pero pueden influir significativamente en el balance final. El efecto hidrófobo es seguro que estabiliza la estructura nativa.
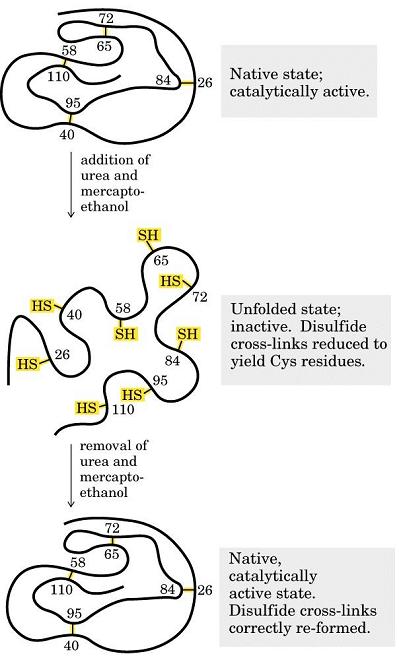
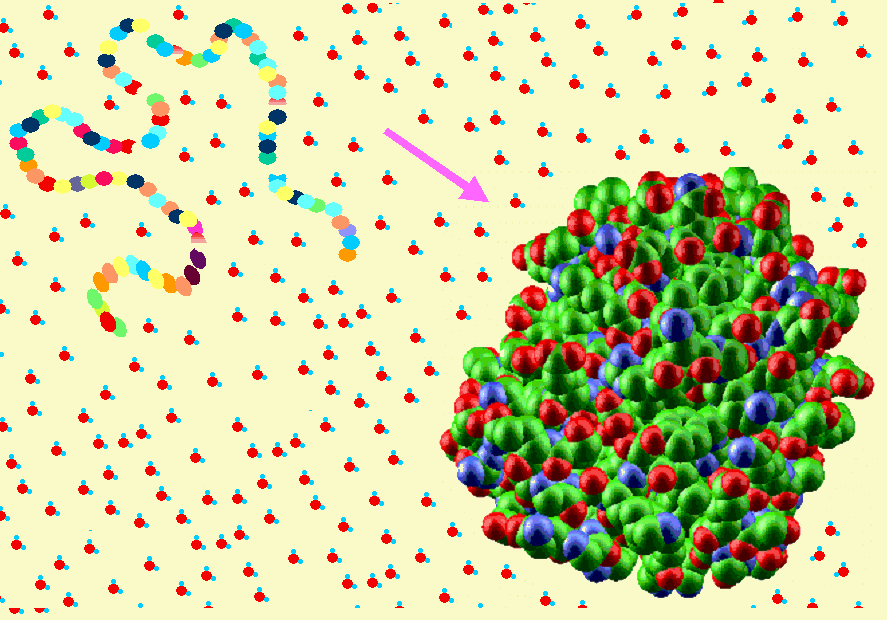
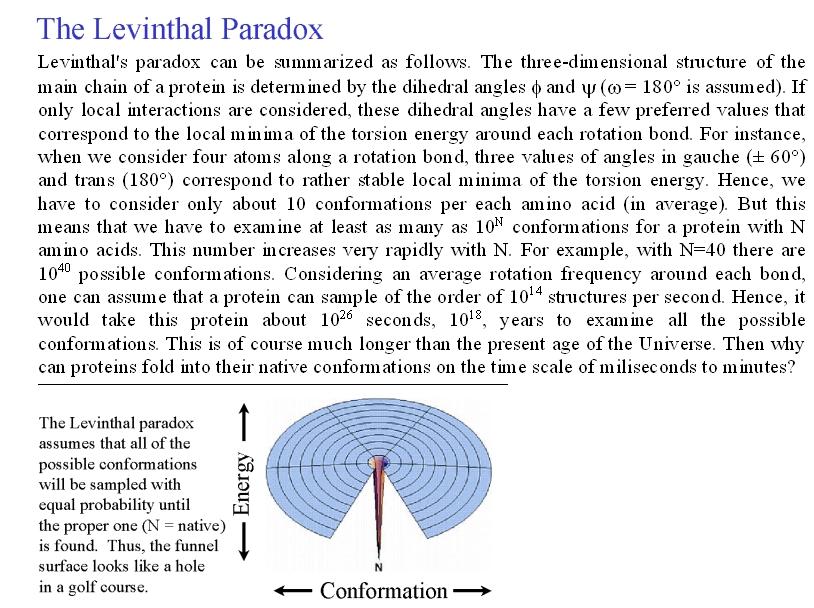
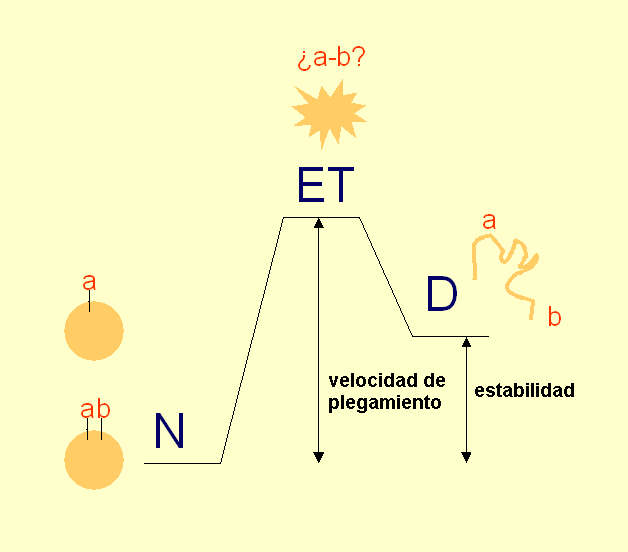
- Experimento de Anfinsen y paradoja de Levinthal. Anfinsen demostró a finales de los 60′ que al desplegar la enzima ribonucleasa A con urea y mercaptoetanol aumentaba su volumen aparente y desaparecían sus propiedades catalíticas. Al dializar la proteína volvía a plegarse. El plegamiento de las proteínas no está inducido por la célula sino que es el resultado de la interacción de la secuencia polipeptídica con el agua. Toda la información necesaria para adquirir su estructura tridimensional está presente en la secuencia de aminoácidos por lo que algún día se podrá predecir. Dada la flexibilidad de los polipéptidos, el número de conformaciones posible de una proteína es enorme. Levinthal planteó la paradoja que lleva su nombre: si una proteína se pliega explorando al azar todas las conformaciones posibles tardará mucho más que la edad que tiene el Universo. Como las proteínas se pliegan muy deprisa (típicamente en milisegundos o segundos) está claro que no exploran todas las conformaciones al azar.
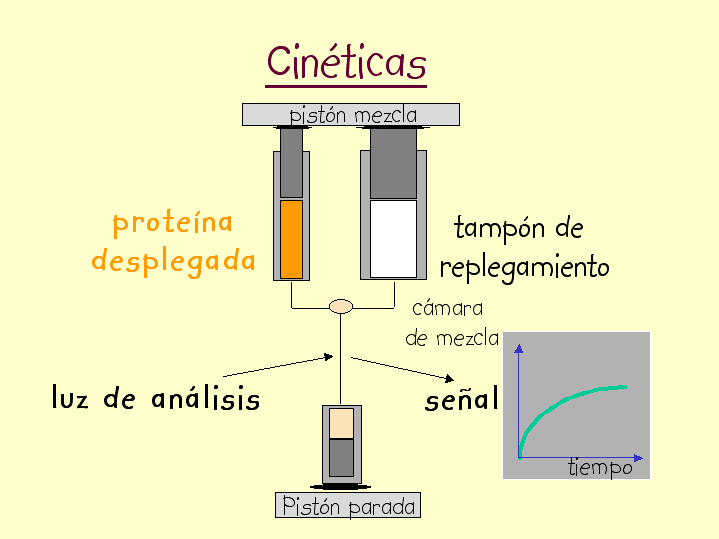
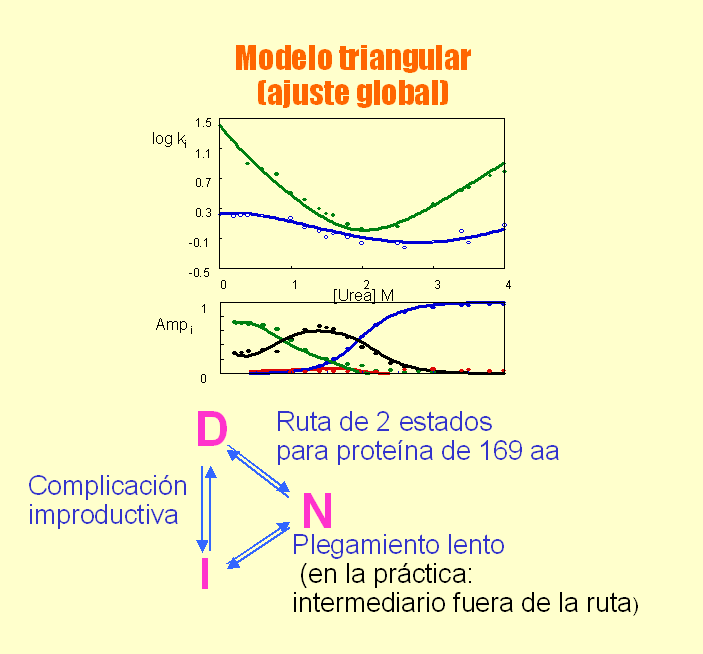
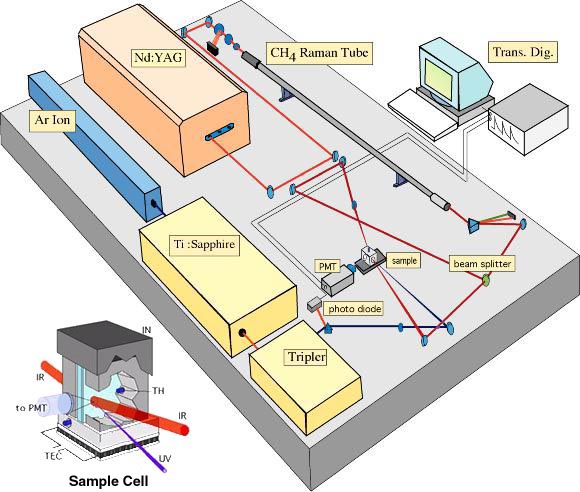
- Medida de la velocidad de plegamiento. Las reacciones muy rápidas no se pueden medir mezclando «a mano» los reactivos. La técnica apropiada para medir reacciones que duran entre decenas de ms y algunos segundos es el flujo detenido stopped-flow. Hay que registrar la evolución en el tiempo de una propiedad espectroscópica (fluorescencia DC en el UV lejano) que distinga al estado desplegado del plegado. El ajuste a una exponencial simple nos permite conocer la constante de la reacción. Si la curva se ajusta a una exponencial doble indica que hay intermediarios en la reacción y es preciso realizar un ajuste global de curvas obtenidas en distintas condiciones para calcular las constantes de velocidad. Muchas proteínas pequeñas se pliegan sin que aparezcan intermediarios (plegamiento de dos estados). Algunos fenómenos tempranos de la reacción de plegamiento ocurren por debajo del milisegundo y requieren otras técnicas como la mezcla en chorro o el salto de temperatura inducido por láser para poderlos observar.
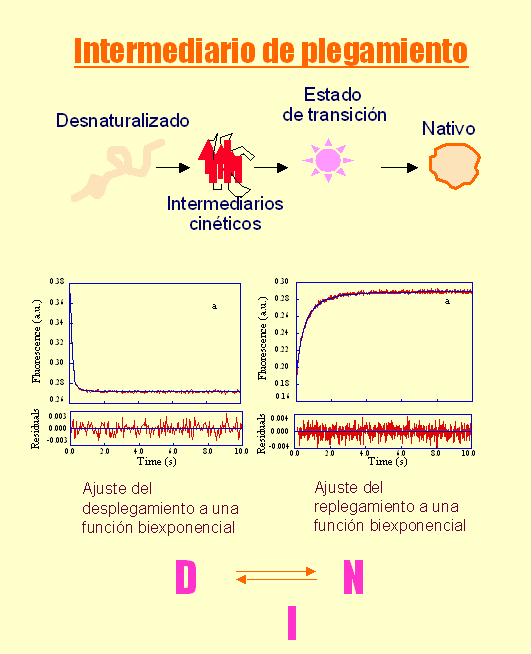
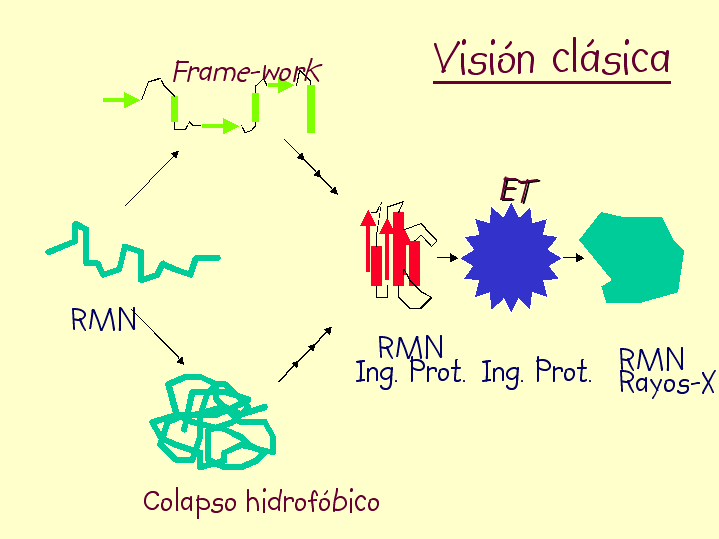
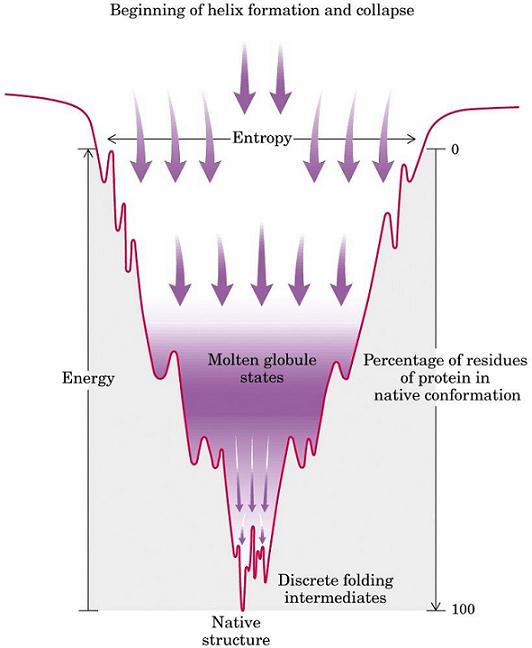
- Modelos clásicos del plegamiento y especies observadas La visión clásica supone que todas las moléculas de proteína siguen el mismo camino pasando por los mismos intermediarios y por el estado de transición. Hay dos clases principales de modelos. Los modelos de tipo armazón (frame-work) asumen que inicialmente se forman algunos de los elementos de estructura secundaria que aparecen en el estado final. Estas regiones chocan y dan lugar a intermediarios que terminan plegándose. El modelo del colapso hidrófobo supone que el primer suceso relevante es un colapso al azar del polipéptido para ocultar los residuos hidrófobos. A partir del estado colapsado se va organizando el polipéptido y aparece la estructura secundaria de los intermediarios y, finalmente, del estado nativo. Experimentalmente las especies que podemos observar y estudiar con más o menos detalle son las siguientes:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
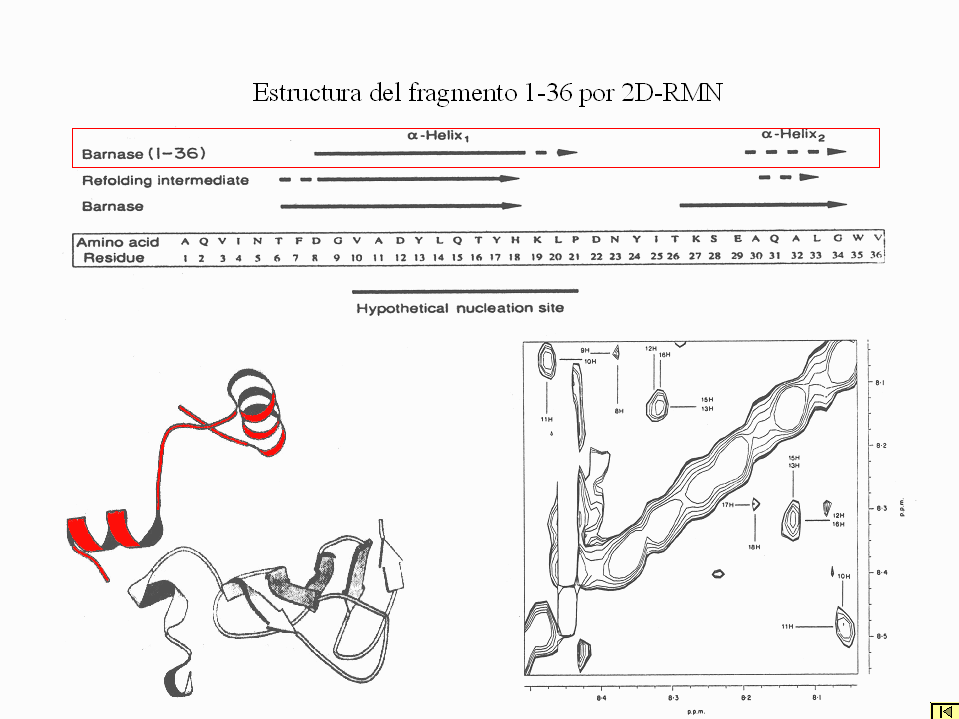
- estado desplegado: se puede estudiar por rmn, con dificultad porque las señales de los residuos del mismo tipo se solapan. Se utilizan también fragmentos de la proteína que simulan su situación en los primeros momentos del plegamiento cuando los residuos del segmento estudiado todavía no han tenido tiempo de interaccionar con el resto de la cadena. Algunas secuencias ya se pliegan aunque débilmente, como lo hacen luego en el estado nativo.
- intermediarios: se pueden estudiar por rmn preparando con aparatos de mezcla rápida muestras de proteína que se incuban durante distintos tiempos (muy cortos) en H2O y luego se transfieren a D2O. Las regiones que ya se habían plegado cuando la proteína se transfiere a D2O retienen los protones fácilmente intercambiables con el disolvente. En general los intermediarios del plegamiento estudiados se parecen ya al estado nativo (aunque con partes desestructuradas y las demás menos consolidadas.
- estado de transición: es la conformación de alta energía por la que atraviesa el polipéptido para plegarse. Sólo se puede estudiar por el método de comparar la velocidad de plegamiento de muchos mutantes distintos. En proteínas pequeñas puede estar limitado a una región pequeña (núcleo de plegamiento). En proteínas de más de 100 residuos suele parecerse bastante a una forma distorsionada del estado nativo.
- estado nativo: su estructura se determina precisamente por rmn o rayos x.
{kind=link}
{kind=link}
Los modelos clásicos secuenciales fueron cuestionados y se propuso una “nueva visión” del plegamiento en la que cada molécula se pliega por una ruta distinta. La razón de que el plegamiento sea rápido es que las moléculas se mueven por un paisaje de energía en forma de embudo de modo que las interacciones nativas que aparecen tienden a conservarse y la proteína sigue plegándose “cuesta abajo”. En general las barreras de energía entre conformaciones son bajas pero donde el embudo presenta rugosidades aparecen intermediarios.
{kind=link}
{kind=link}
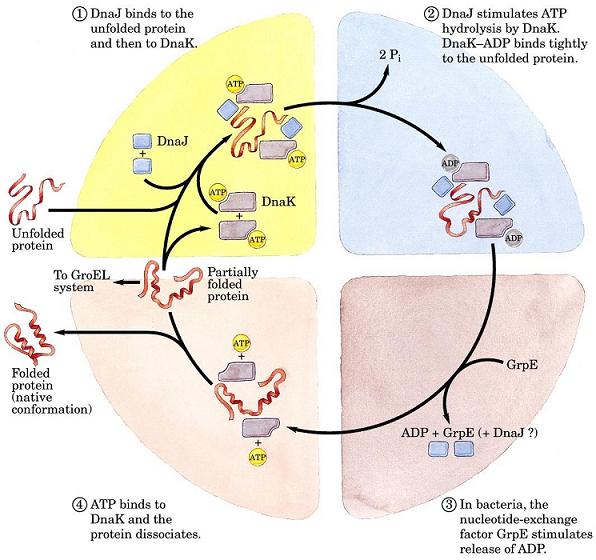
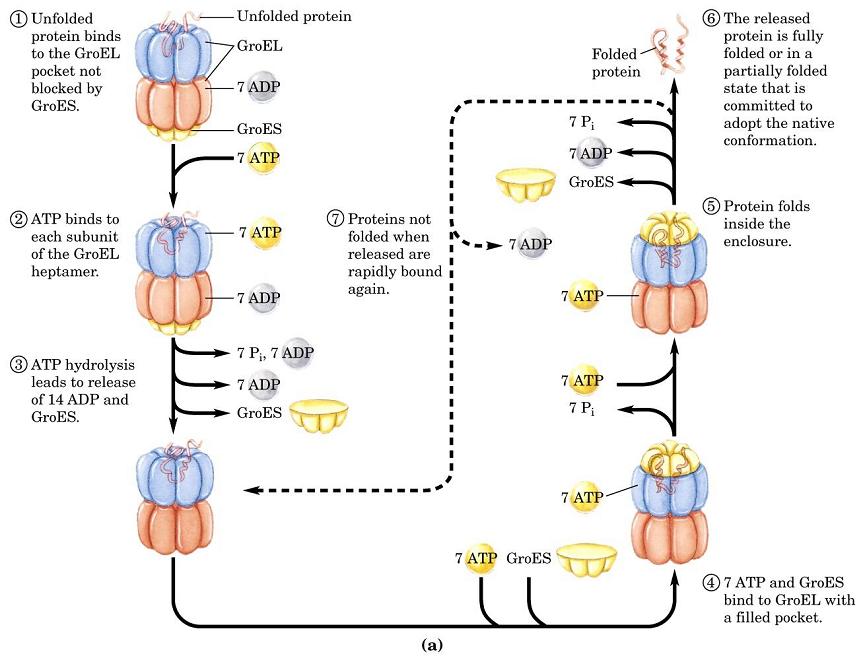
- Plegamiento in vivo y chaperonas. Aunque toda la información que precisa una proteína para plegarse reside en su propia secuencia, el interior de la célula está tan repleto de proteínas que aparecen problemas de agregación, pues los intermediarios del plegamiento pueden tener expuestas regiones hidrófobas que son «pegajosas». Para evitar estos problemas o los que surgen como consecuencia de un estrés térmico (intermediarios parcialmente desnaturalizados con el mismo problema de tendencia a agregar) la célula expresa proteínas auxiliares: chaperonas que secuestran temporalmente las conformaciones no plegadas, o chaperoninas que desnaturalizan a las mal plegadas y les dan otra oportunidad de plegarse correctamente.
{kind=link}
{kind=link}
{kind=link}