
The Nobel Prize in Chemistry 1962 Max Ferdinand Perutz and John Cowdery Kendrew
"For their studies of the structures of globular proteins"
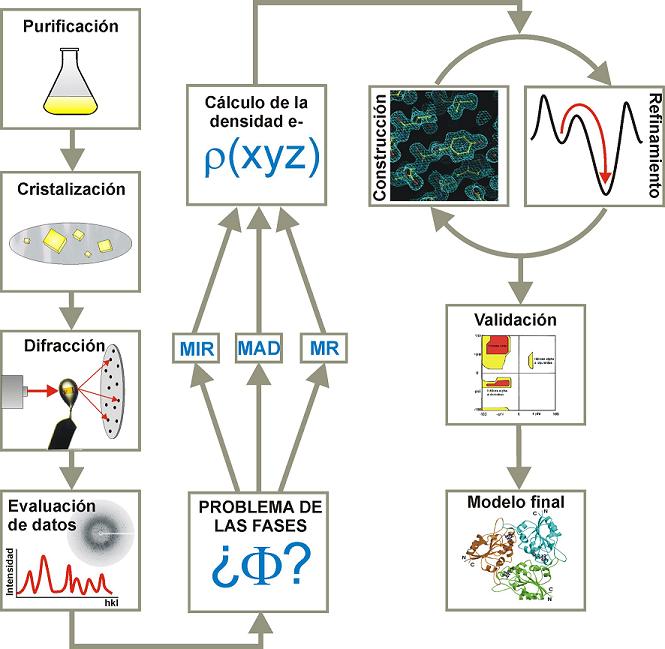
- Los rayos X. Los rayos X son radiación electromagnética de longitud de onda corta (en el rango del Ă). 1895-ROENTGEN descubre que, cuando un haz de e– rápidos golpea una superficie sólida, se emite radiación que atraviesa la materia e impresiona la película fotográfica. A los tres meses se utilizan ya en cirugía. 1913-Se resuelve la estructura del cristal de NaCl. 1959-PERUTZ y KENDREW resuelven las estructuras proteicas de la hemoglobina y la mioglobina. 2003-Más de 18.000 estructuras proteicas resueltas por rayos X.
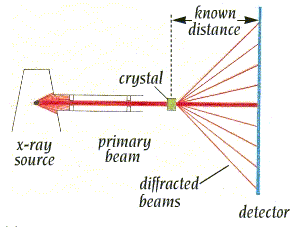
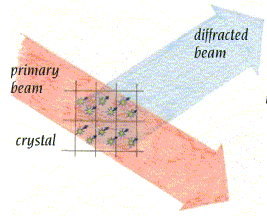
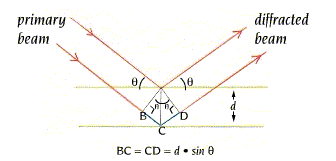
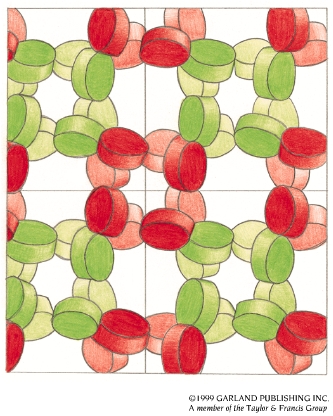
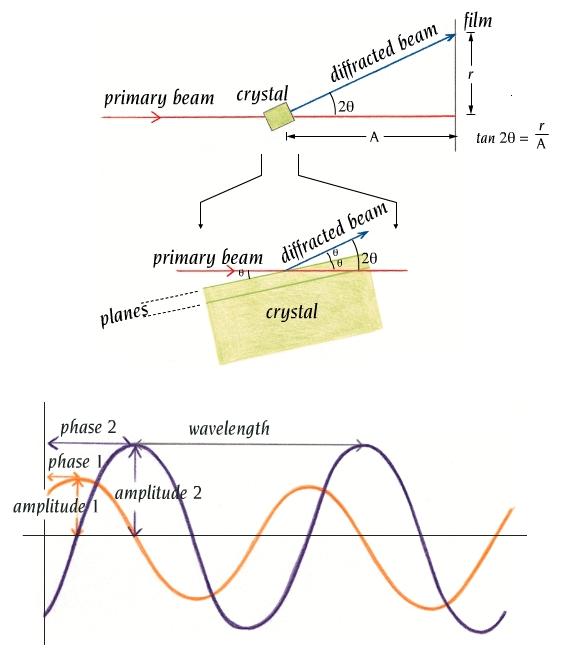
- La difracción de rayos X por un cristal. Cuando un haz de rayos X incide en un cristal, parte de los rayos lo atraviesan y parte son dispersados por los e–de los átomos del cristal. La mayor parte de los fotones dispersados se cancelan entre si (sus ondas interfieren y se anulan). En ciertas direcciones, sin embargo, los fotones salen en fase y sus ondas se refuerzan dando lugar a un haz de rayos X difractados. Las direcciones en que los fotones se refuerzan están determinadas por la ley de Bragg (2dsenθ=nλ): si el desfase es múltiplo de la longitud de onda, los fotones siguen en fase. Cualquier conjunto de planos de un cristal orientados respecto a la fuente de rayos X de modo que se cumpla la ley de Bragg son capaces de ‘reflejar’ fotones en fase y dar lugar a un haz difractado. En un cristal hay diferentes conjuntos de planos paralelos que cumplen la ley de Bragg. El conjunto de haces difractados por un cristal constituye su patrón de difracción. El patrón de difracción es la clave para reconstruir la estructura de la proteína.
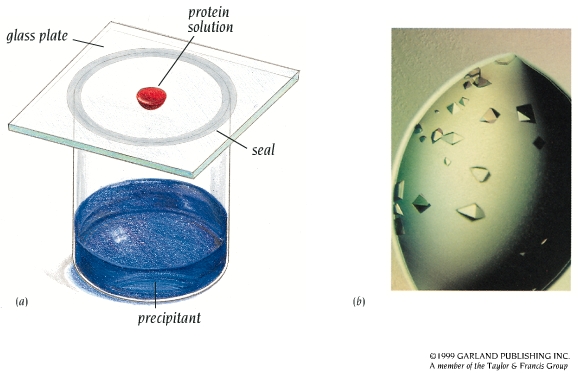
- Los cristales de proteínas. Se obtienen a partir de soluciones concentradas de proteína pura. Un método habitual es el de la gota colgante. La cristalización es muy sensible a la temperatura, pH, fuerza iónica, y a la presencia de otras moléculas en la disolución. Hay que probar muchas condiciones hasta acertar. Hay robots y ‘kits’ comerciales que ayudan a encontrar las condiciones adecuadas. Los cristales de proteínas contienen aproximadamente un 50 % de disolvente (entre 25-90 %). Esto tiene una ventaja (están ‘casi en disolución’ y su estructura no se altera) y un inconveniente (son frágiles).
- La toma de datos
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
-
- Tipos de fuente. Rayos X monocromáticos: Electrones acelerados golpean un ánodo metálico que emite rayos X. Hay que ir girando el cristal para variar el ángulo θ y conseguir nuevas series de planos que cumplan la ley de Bragg y generen nuevos haces difractados. Rayos X policromáticos: Electrones a velocidades próximas a la velocidad de la luz que proceden de un sincrotrón. Aunque al hacerlos monocromáticos pierden intensidad, la que queda es muy superior a la de las fuentes convencionales y con ellos se acorta mucho el tiempo de adquisición.
- Tipos de detector. Detector electrónico de área que almacena automáticamente en un ordenador los ángulos e intensidades de cada haz difractado. Antiguamente se utilizaba película fotográfica, regla, etc.
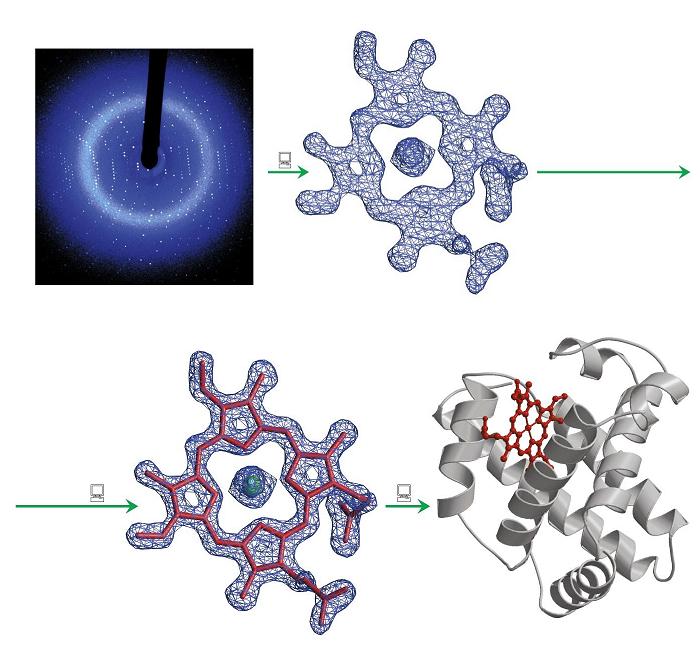
- El mapa de densidad electrónica de la proteína. Cada una de las manchas del patrón de difracción de un cristal está producida por un haz de difracción al que contribuyen todos átomos del cristal. El haz difractado es una onda cuya ecuación se puede rescribir en función de las coordenadas de cada átomo que ha contribuido a generarla:
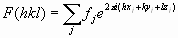 . La transformada de Fourier permite rescribir la ecuación como:
. La transformada de Fourier permite rescribir la ecuación como: 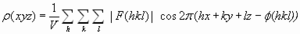 , que da el mapa de densidad electrónica: la representación tridimensional de la densidad electrónica de la molécula. Este mapa es la herramienta fundamental que permite deducir la estructura de la molécula. Para calcularlo hay un problema: el patrón de difracción permite medir la intensidad de cada haz pero no su fase (problema de la fase). Soluciones al problema de la fase: Para moléculas pequeñas: Se inventan fases y se calcula el mapa que se compara con la molécula. Si no es razonable se rechazan las fases y se intenta con otras. Para proteínas:(a) remplazamiento isomorfo Se derivatizan los cristales con átomos pesados (Hg, I, Se,…). Se comparan los patrones de difracción del cristal original con los de 2 cristales derivatizados distintos. De la comparación de los 3 cristales se calculan las fases. (b) remplazamiento molecular. Se utilizan las fases de una proteína de estructura parecida y conocida. (c) dispersión anómala. Se obtiene proteína con metioninas sustituidas por selenometioninas y se aprovechan las propiedades de dispersión del Se (que químicamente se comporta parecido al azufre). Una vez conocidas las amplitudes y las fases, la transformada de Fourier calcula el mapa de densidad electrónica.
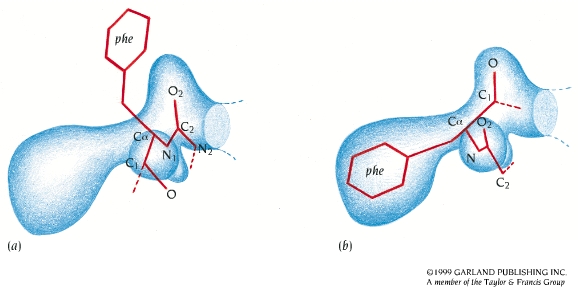
, que da el mapa de densidad electrónica: la representación tridimensional de la densidad electrónica de la molécula. Este mapa es la herramienta fundamental que permite deducir la estructura de la molécula. Para calcularlo hay un problema: el patrón de difracción permite medir la intensidad de cada haz pero no su fase (problema de la fase). Soluciones al problema de la fase: Para moléculas pequeñas: Se inventan fases y se calcula el mapa que se compara con la molécula. Si no es razonable se rechazan las fases y se intenta con otras. Para proteínas:(a) remplazamiento isomorfo Se derivatizan los cristales con átomos pesados (Hg, I, Se,…). Se comparan los patrones de difracción del cristal original con los de 2 cristales derivatizados distintos. De la comparación de los 3 cristales se calculan las fases. (b) remplazamiento molecular. Se utilizan las fases de una proteína de estructura parecida y conocida. (c) dispersión anómala. Se obtiene proteína con metioninas sustituidas por selenometioninas y se aprovechan las propiedades de dispersión del Se (que químicamente se comporta parecido al azufre). Una vez conocidas las amplitudes y las fases, la transformada de Fourier calcula el mapa de densidad electrónica. - Ajuste de la secuencia de aminoácidos al mapa. Cuanto más precisamente se han determinado amplitudes y fases, cuanto más ordenado es el cristal y cuantas más reflexiones se han utilizado en los cálculos, más se parece el mapa de densidad electrónica a la estructura de la molécula. Es necesario conocer la secuencia de la proteína (o del polinucleótido, en su caso) para ajustarla al mapa de densidad electrónica y construir el modelo inicial. Este ajuste es en parte automático y en parte ‘manual’. Lo más importante es que las fases estén bien.
- Refinamiento. Al modelo realizado se le calcula el patrón de difracción a que daría lugar y se compara con el observado mediante el parámetro R. Un modelo absurdo daría R ~0.59. El modelo es aceptable a partir de R<0.2. Con proteínas se puede llegar hasta R = 0.1 y con moléculas pequeñas hasta R = 0.01. Para refinar el modelo: se incluyen moléculas de agua, se varía el factor de temperatura de cada átomo (es una medida de su movilidad), se varían ligeramente las fases, se varían las coordenadas (‘manualmente’ o por dinámica molecular)
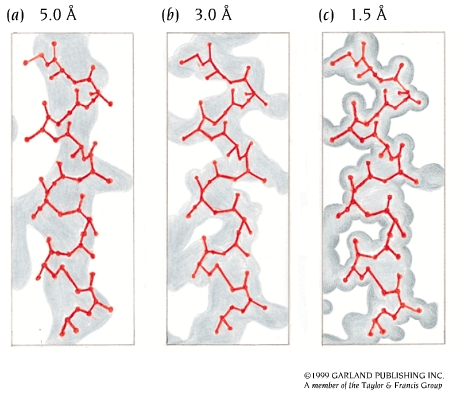
En cristalografía de proteínas se denomina resolución a la distancia menor entre planos refractantes para la que se ha podido recoger reflexiones (cuanto menor es la resolución, mayor es el número de reflexiones utilizadas). Son resoluciones aceptables entre 1.5 y 2.0 Å, lo que equivale a una imprecisión en las coordenadas de ~ 0.15 Å.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
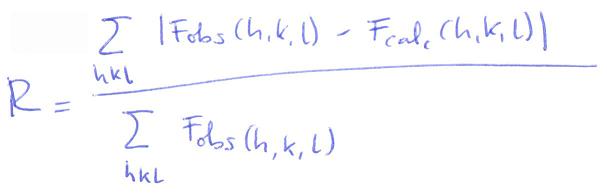{kind=link}