- El spín nuclear y la resonancia magnética nuclear
- El desplazamiento químico
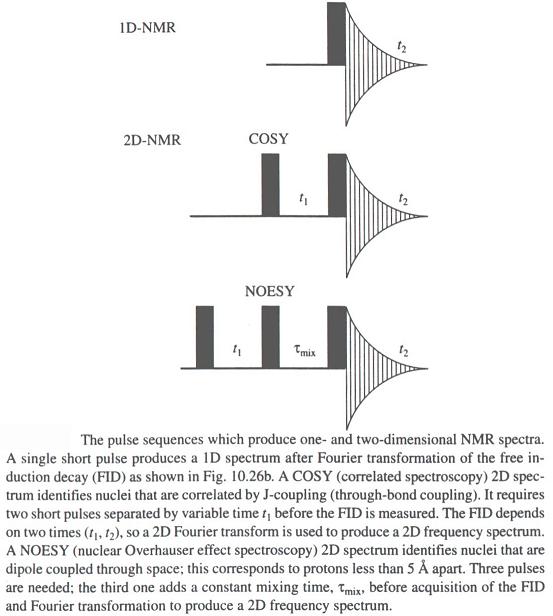
- RMN monodimensional
- Acoplamiento de espines
- El efecto Overhauser nuclear (NOE)
- RMN bidimensional; espectros COSY de los aminoácidos
- Asignación de los sistemas de spin
- El espectros NOESY; asignación de la secuencia
- Los NOEs no secuenciales; calculo de estructuras compatibles
- Marcación con 13C y con 15N; RMN multidimensional
- RMN versus Rayos X.

The Nobel Prize in Chemistry 1991 Richard R. Ernst
"For his contributions to the development of the methodology of high resolution nuclear magnetic resonance (NMR) spectroscopy"

The Nobel Prize in Chemistry 2002 Kurt Wüthrich
"For his development of nuclear magnetic resonance spectroscopy for determining the three-dimensional structure of biological macromolecules in solution"

The Nobel Prize in Physiology or Medicine 2003 Paul C. Lauterbur and Sir Peter Mansfield
"For their discoveries concerningmagnetic resonance imaging"
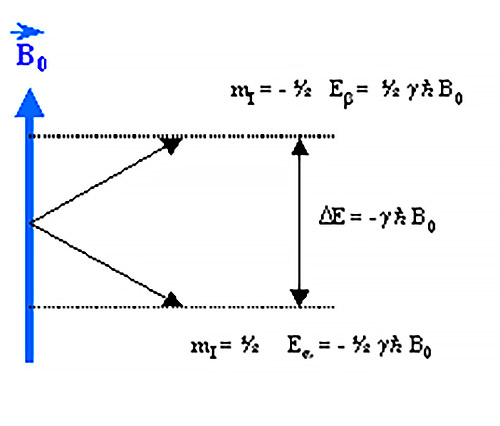
- Elspínnuclear y la resonancia magnética nuclear. Los núcleos de muchos elementos químicos poseen momentos dipolares magnéticos caracterizados por un número cuántico de spin: I. Según el núcleo, I puede valer 0 (12C, 16O, 32S), ½ (1H, 13C, 15N, 19F, 31P), 1 (14N). En presencia de un campo magnético los núcleos se pueden encontrar en 2I+1 niveles de energía distintos. Para el protón (núcleo de 1H) hay 2 x ½ + 1 = 2 niveles de energía. Un protón en el nivel inferior puede absorber radiación electromagnética (ondas de radio) de frecuencia υ0 y pasar al estado superior. El protón que absorbe energía se dice que está en resonancia con el campo electromagnético.
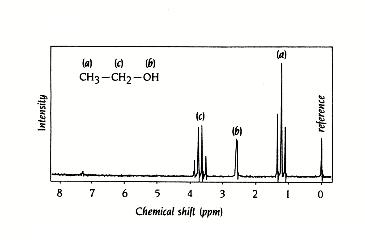
- El desplazamiento químico. Al aplicar un campo magnético a una molécula se inducen momentos magnéticos electrónicos de sentido contrario al campo aplicado que lo «apantallan» ligeramente. El grado de apantallamiento (definido por la constante de apantallamiento: σ) depende de la densidad electrónica de cada región de la molécula. Según el apantallamiento que experimenta cada protón su frecuencia de resonancia es ligeramente distinta: hν = 2mmH0(1- σ). Utilizando una sustancia de referencia, se define el desplazamiento químico de cualquier protón como: δ (ppm) =106(νmuestra – νreferencia)/ νespectrómetro. El desplazamiento químico de un protón es independiente del instrumento utilizado y tiene valores característicos para cada grupo funcional.
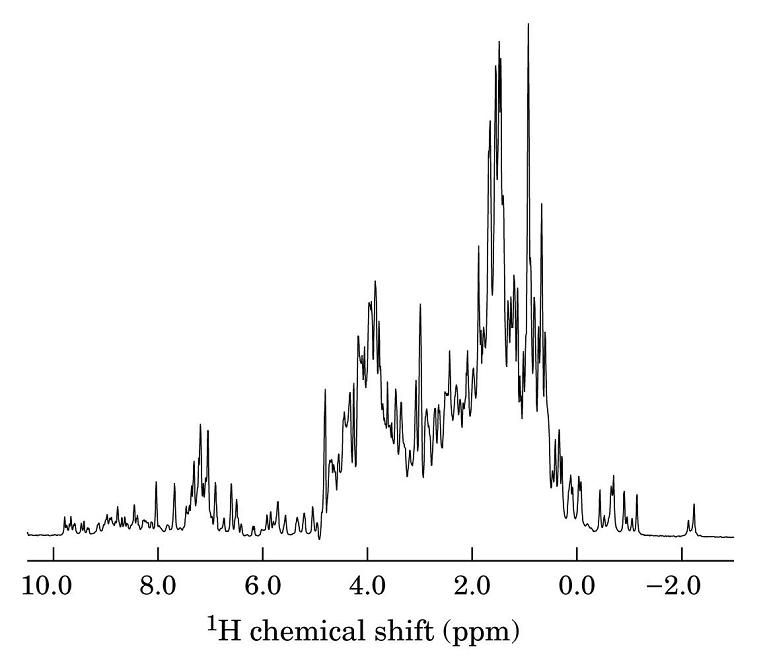
- RMN monodimensional
{kind=link}
-
- Método antiguo: Se varía la frecuencia de la radiación electromagnética y se registra la absorción de radiación.
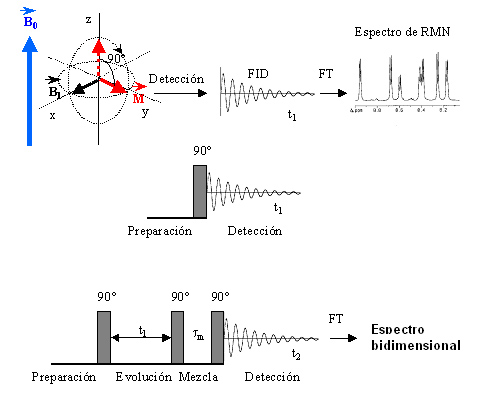
- Método actual: Se lanza un pulso corto (ms) de radiofrecuencia que contiene simultáneamente todas las frecuencias de excitación. Los protones absorben la radiación y se orientan generando un nuevo campo magnético que se puede medir así como su decaimiento temporal («free induction decay»: FID). A esta señal temporal, S(t), se le aplica una transformada de Fourier que la convierte en intensidad absorbida en función de la frecuencia de la radiación de excitación I(n). Cada protón absorbe a una ν distinta y sus decaimientos se recogen superpuestos. La transformada recompone el espectro en función de la frecuencia.
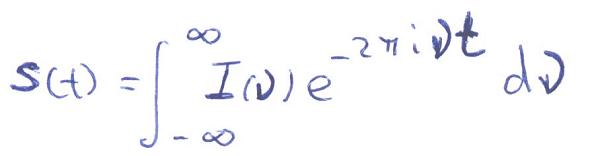{kind=link}
{kind=link}
{kind=link}
- Acoplamiento de espines. El campo magnético de un protón influye en el de los protones vecinos. El campo magnético y la frecuencia de absorción de un determinado protón es: ν = 2mmHo/h. Un protón vecino, modifica ligeramente Ho haciéndolo mayor Ho+ o menor Ho– de modo que el protón puede absorber a ν´ = 2mmHo+/h ó a ν´´ = 2mmHo–/h. Así la señal se desdobla y los dos subpicos se separan según la constante de apantallamiento: J = (ν´-ν´´). Los protones acoplados de esta manera son los separados mediante 3 enlaces químicos.
- El efecto Overhauser nuclear (NOE). Un protón, cuya proporción de núcleos con spin +1/2 y –1/2 está alterada (se consigue con una fuerte irradiación del protón) interactúa por acoplamiento dipolar con un protón vecino, alterando su proporción de núcleos con spin +1/2 y –1/2 y cambiando así la intensidad de la absorción. El desplazamiento químico no se altera. Se utiliza para detectar los protones que están cerca de otros. Es muy sensible a la distancia porque la interacción dipolar depende de r-6. Sólo actúa a distancias <5Å y no importa el número de enlaces químicos que separan a los dos protones.
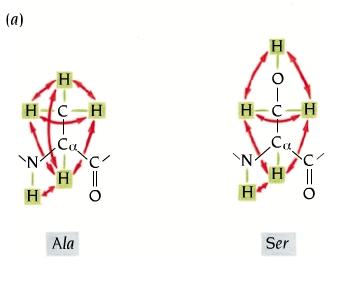
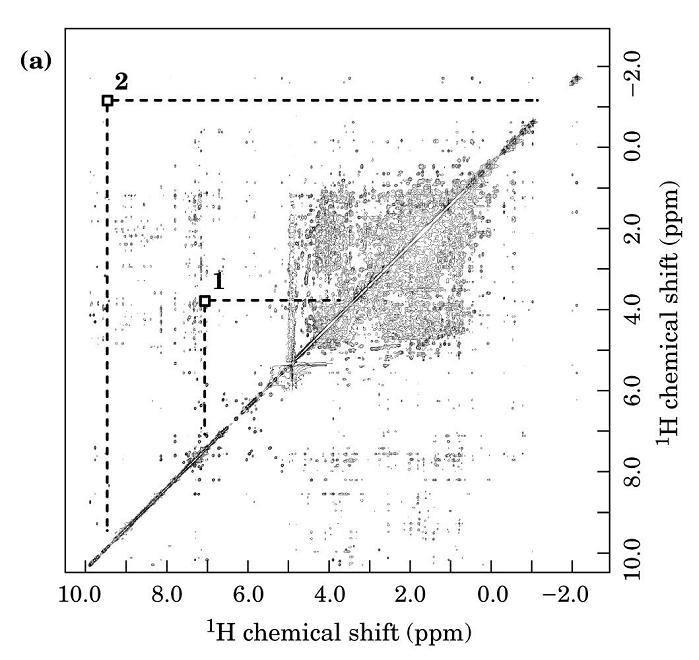
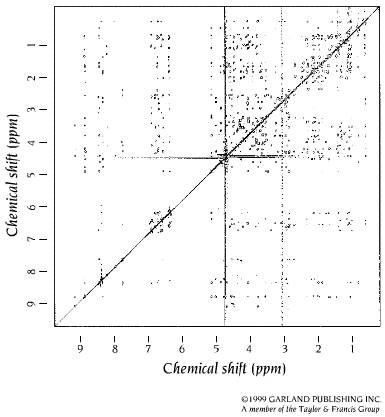
- RMN bidimensional; espectros COSY de los aminoácidos. El espectro de rmn de un proteína pequeña es ya demasiado complejo (18kD ~1500 protones). Para identificar el pico correspondiente a cada protón es necesario dispersarlos en, el menos, dos dimensiones. Esto se consigue mediante secuencias de pulsos de radiofrecuencias con dos variables temporales. La técnica más común es el espectro COSY en el que se registran FIDs con distintos t1. La transformada de Fourier recompone el espectro COSY bidimensional. La diagonal es el espectro monodimensional y lo interesante son los picos que están fuera de la diagonal. Las coordenadas de cada uno de dichos picos son los desplazamientos químicos de dos protones con acoplamiento de spines (que están separados por 3 enlaces químicos). Los picos corresponden a los protones de los distintos aminoácidos de la proteína. Cada aminoácido da un conjunto de picos característico. Algunos aminoácidos dan un patrón COSY idéntico (C, D, N, S, H, F, Y, W), (E, Q, M) mientras que otros poseen patrones específicos: G,A,I,L,V,P,K,R,T.
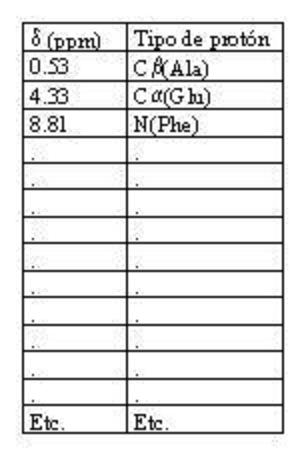
- Asignación de los sistemas de spin. El primer paso es distribuir todos los picos del espectro COSY en patrones como los anteriores. Así el espectro se convierte en un conjunto de patrones de picos (cada uno correspondiente a un tipo de aminoácidos. Una vez asignados todos los picos a alguna clase de aminoácido se genera una tabla. En el espectro COSY, los protones de cada residuo están aislados de los de residuos adyacentes de modo que el espectro COSY no da información de cómo se ordenan los aa en la proteína.
- El espectro NOESY; asignación de la secuencia. Se obtiene con una secuencia de pulsos aún más compleja. Cada pico fuera de la diagonal (NOE) tiene por coordenadas los desplazamientos químicos de un par protones cercanos en el espacio (con independencia de que estén cercanos en la secuencia). El protón NH de un residuo i siempre queda cerca del protón de C alfa o de C beta del i-1 dando lugar a picos intensos cuyas coordenadas permiten saber (consultando la tabla) que a un determinado tipo de aa (por ej: Tyr) le precede otro de un tipo determinado (por ej: Thr). Así se sabe, en este ej., que los desplazamientos del pico corresponden a una secuencia Thr-Tyr. A continuación se busca en la tabla el desplazamiento químico del NH de esa Thr y se busca en el espectro NOESY el pico que da con el C alfa del residuo que la precede (i-2) (por ej: Phe). Así se determina que los desplazamientos de los tres aa en cuestión corresponden a los de la secuencia Phe-Thr-Tyr. Cuando se tiene una secuencia suficientemente larga se compara con la de la proteína para establecer a qué aa de la secuencia global corresponden. De este modo, en la tabla de asignaciones, a cada uno de los aminoácidos se le pone su número y así, cada desplazamiento químico del espectro queda asignado específicamente a uno de los protones de la proteína.
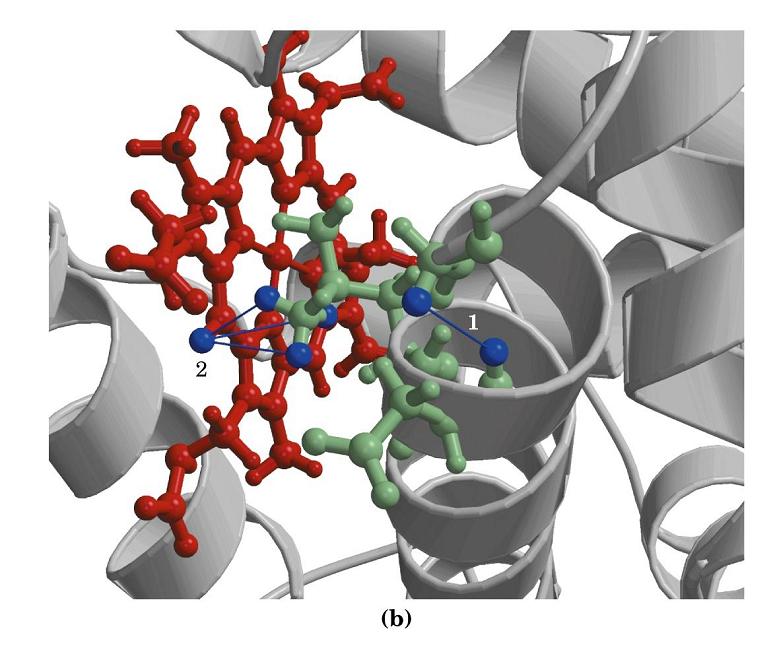
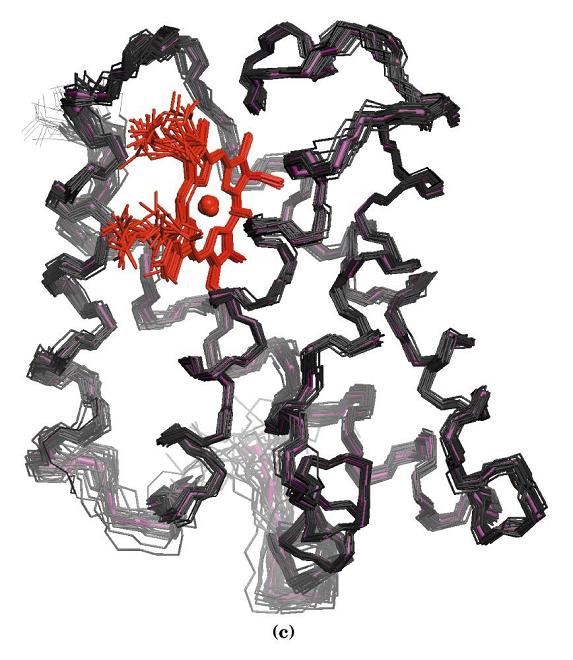
- Los NOEs no secuenciales; cálculo de estructuras compatibles. Cada NOE no secuencial tiene por coordenadas los desplazamientos químicos de dos protones (la tabla de asignación nos dice a qué aminoácidos corresponden) que están cerca en el espacio ( < 5 Å) La lista de NOEs no secuenciales dice qué aa están cerca de otros en la estructura terciaria de la proteína. Un programa de ordenador calcula estructuras compatibles con los NOEs observados (cada NOE restringe el número de estructuras posibles). Si el número de NOEs es suficiente, se puede determinar la estructura tridimensional de una proteína con buena resolución .
- Marcación con 13C y con 15N; RMN multidimensional. En proteínas de cierto tamaño, dispersar los picos del espectro en dos dimensiones no es suficiente para separarlos bien y poder asignarlos. En estos casos hay que obtener muestras de proteína enriquecidas en 13C y/o 15N y utilizar secuencias de pulsos más complejas para dispersar la señal en 3 y hasta en 4 dimensiones. La proteína marcada se obtiene como proteína recombinante cultivando el organismo que contiene el gen en un medio que carezca de carbono (y/o nitrógeno) suplementado con 13C-glucosa y/o 15NH4Cl.
- RMN versus Rayos X. La rmn se realiza en solución acuosa, requiere una alta solubilidad de la proteína (1 mM), permite el estudio de procesos dinámicos y está limitada normalmente a proteínas de menos de 100 kD. La difracción de rayos x se realiza en un cristal (la proteína no tiene que ser tan soluble pero tiene que cristalizar), apenas permite el estudio de procesos dinámicos y no está limitada a proteínas pequeñas.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}